本文共 1158 字,大约阅读时间需要 3 分钟。
mTM-aligin: an algorithm for fast and accurate multiple protein structure alignment
蛋白结构在进化中更加保守,因此根据多个结构进行比对比仅仅依赖于多重序列联配(MSA)更加有意义,特别是那些远房亲戚。作者看着PDB(protein data bank)那日益增加的蛋白结构心生忧愁,那么多数据不开发一个有效的比对算法是吃枣药丸的。
于是他写了一个mTM-algin, 是TM-algin的扩展(谁能告诉我什么叫做TM-align),他还带着这个算法去HOMSTRAD, SABmark_sup, SABmark_twi, SISY-multiple这些数据库溜达了一圈,非常开心的说了一句,我的算法比你们表现的更好哦。 工具地址:BioSeq-Analysis: a platform for DNA, RNA and protein sequence analysis based on machine learning approaches
我们都知道当测序测得七七八八之后,就是后基因组学的时代,那么最大的挑战就是如何解读这个无字天书,了解他们的结构和功能。机器学习(ML, make...)是当今最火的领域之一,隔壁还有一家深度学习,肯定要到生物大数据里面玩一下,一般步骤就是:特征提取,构建预测模型,性能评估。作者说了,尽管目前已经有很多在线或离线(stand-alone)工具被开发出来,但是都局限于其中的一步而已。于是作者就做了一个微小的工作,开发了BioSeq-Analysis能够自动化实现以上三步,也就是能够根据评估数据集自动产生最优预测模型,并且报告性能,更加厉害的是他还支持Windows,Linux,Unix哦
工具下载地址:不是我吐槽,这个网页也。。。
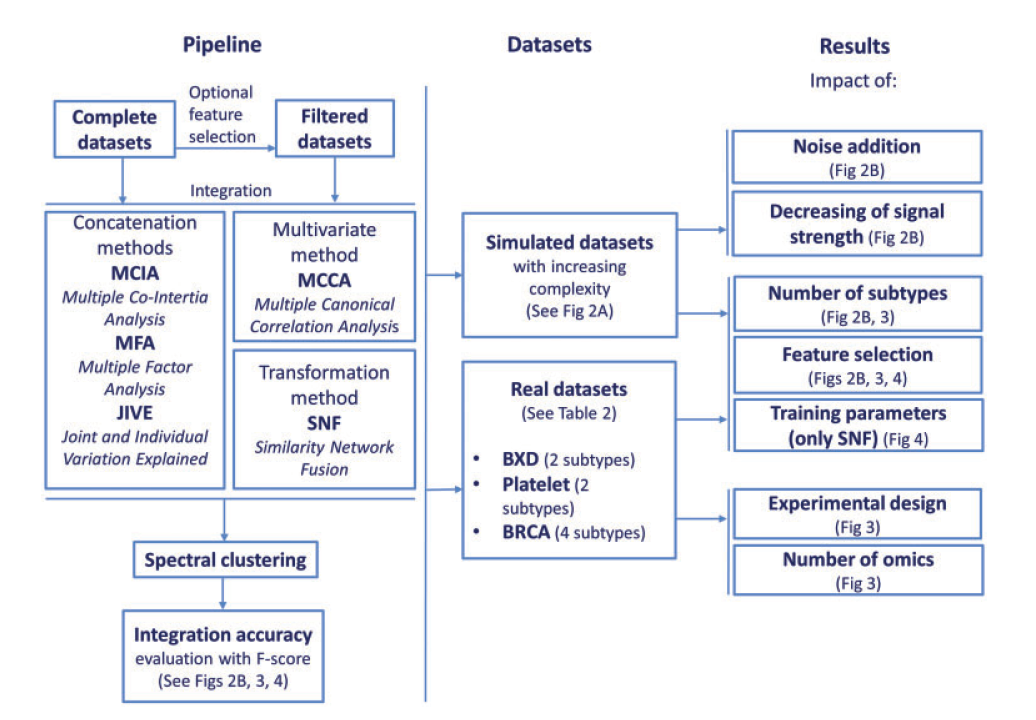
Multi-omics integration—a comparison of unsupervised clustering methodologies
多组学(multi-omics)目前发展很快,分析过程存在很多因子影响着结果的准确性,比如说样本聚类。作者比较多个无监督聚类方法:Multiple Canonical Correlation Analysis, Multiple Co-Inertia Analysis, Multiple Factor Analysis, Joint and Individual Variation Explained,Similarity Network Fusion。采用已经报道文献的标准数据集,分别评估不同算法在实验设计,特征选择和参数训练等方面的差异。
没有涉及这个领域,表示看不懂呀。
转载地址:http://utexa.baihongyu.com/